
Rétinite pigmentaire : symptômes, âge, qu'est-ce que c'est ?

Cette maladie génétique affecte la rétine et évolue vers la perte de la vue.

Peu connue, la rétinite pigmentaire est une maladie de la vue qui touche la rétine et entraîne une cécité. Elle touche indifféremment les femmes et les hommes, quelle que soit leur origine géographique. À quoi est-elle due ? À quel âge apparaît-elle et sous quelles formes ? Quels sont les traitements ? Les docteurs Chafik Keilani et Raphaël Atia, chirurgiens ophtalmologistes nous expliquent cette maladie.
Définition : c'est quoi la rétinite pigmentaire ?
La rétinite pigmentaire est une maladie génétique dégénérative de l'œil caractérisée par une perte progressive et graduelle de la vision évoluant généralement vers la cécité. Elle est aussi appelée RP ou rod-cone dystrophie ou retinitis pigmentosa, synonymes dérivés de son nom en anglais.
Quelles sont ses causes ?
La rétinite pigmentaire est une maladie génétique due à la mutation de gènes impliqués dans le fonctionnement des photorécepteurs, cellules de la rétine, indispensables pour la vision.
► "Dans la moitié des cas, la personne atteinte est la première à l'être dans la famille, explique le Dr Kelani. On dit alors que la rétinite pigmentaire est sporadique. Dans ce cas, la maladie est la conséquence d'une mutation génétique survenue inopinément, elle est transmissible à la descendance".
► Dans l'autre moitié des cas, la rétinite pigmentaire est dite "familiale", car au moins deux personnes de la même famille sont atteintes.
Plusieurs gènes entraînant la maladie ont été décrits et la transmission peut se faire selon tous les modes : autosomique dominant, autosomique récessif, transmission liée à l'X. Les gènes responsables sont très nombreux, plus d'une trentaine et la recherche continue d'en découvrir. La mutation de certains gènes est plus fréquente comme celle de la rhodopsine (RHO), du gène retinitis pigmentosa 1 (RP1), du gène RPGR... Cette maladie ne touche donc habituellement que des frères et sœurs dans une même famille. La probabilité d'avoir un autre enfant atteint est d'une sur quatre.
Âge
Le rétinite pigmentaire peut débuter à n'importe quel âge selon le gène malade avec une fréquence d'apparition plus grande entre 10 et 30 ans.
Symptômes : une mauvaise vision la nuit
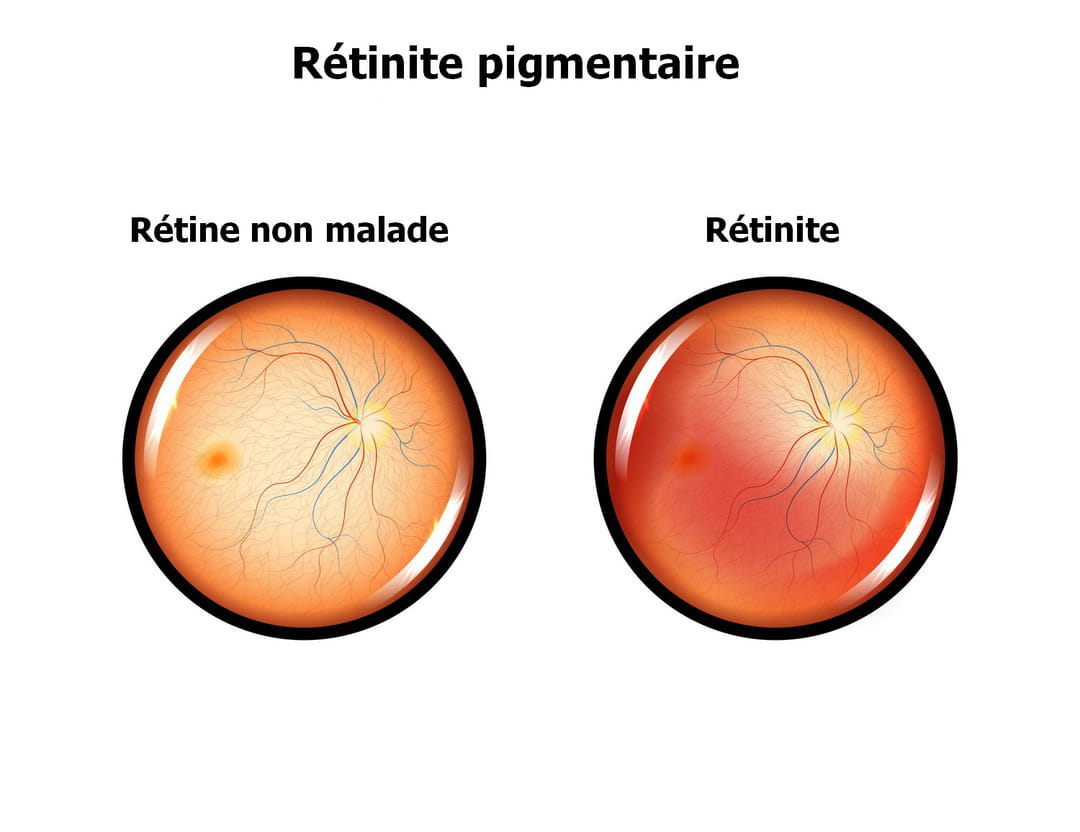
La rétinite pigmentaire débute généralement par des problèmes de vue lorsque l'intensité de la lumière diminue (héméralopie). "La première plainte sera une mauvaise vision la nuit, signale le Dr Kelani. Des difficultés d'adaptation à l'obscurité sont fréquentes". Puis progressivement, le champ visuel se rétrécit avec l'impossibilité de voir les choses en haut, en bas ou sur les côtés, donnant une impression de "vision en tunnel". Cette atteinte est bilatérale, c'est à dire que les deux yeux sont affectés. Cela altère progressivement les gestes de la vie quotidienne : la personne développe une certaine maladresse, des difficultés à conduire de nuit, parfois aussi de jour... Des troubles dans la vision des couleurs sont souvent présents (dyschromatopsie). La vision centrale est généralement conservée jusqu'à des stades tardifs de la maladie. Sa diminution se manifeste d'abord par des difficultés à réaliser des activités minutieuses ou à lire puis, progressivement l'acuité visuelle diminue et aboutit généralement à la cécité. La cataracte, qui est
une opacification du cristallin perturbant progressivement la vision, est également une complication fréquente.
Diagnostic
Le diagnostic est établi lors d'un bilan ophtalmologique effectué suite à des problèmes de vision nocturne, un accident par défaut de vision périphérique, ou une baisse d'acuité visuelle. Des examens complémentaires confirmeront le diagnostic. L'électrorétinogramme permet d'explorer l'activité cellulaire rétinienne au cours d'une stimulation lumineuse et dans le noir ; il met en évidence le dysfonctionnement des cellules de la rétine. La réalisation d'un électrorétinogramme ne nécessite pas d'hospitalisation, il est indolore. L'examen du fond de l'œil après avoir dilaté la pupille avec un collyre met en évidence la présence des dépôts pigmentés sur la rétine. Un examen du champ visuel, central et périphérique, est réalisé et permet d'évaluer le handicap visuel. D'autres examens complémentaires peuvent éventuellement être réalisés. Lorsque le diagnostic de rétinite pigmentaire est posé, le médecin pourra rechercher la mutation d'un gène déjà connu par un test génétique.
Quels traitements ?
"Il n'existe pas, à l'heure actuelle, de traitement permettant de guérir de la rétinite pigmentaire, déplore le Dr. Kelani. Quelques précautions peuvent ralentir la progression de la maladie. Le port de verres protecteurs et filtrants adaptés, protégeant de la luminosité et des rayons ultraviolets est recommandé". Leur but est également de diminuer la sensation d'éblouissement, tout comme le port d'un chapeau. Il est également conseillé d'éviter les expositions au soleil. On peut aussi utiliser des aides pour améliorer la vision comme des lunettes grossissantes, des loupes ou des télescopes. D'autres options incluent les livres à gros caractères et les téléphones adaptés. Des dispositifs de lecture informatisés peuvent aussi aider, tout comme le suivi par une équipe médicale variée, incluant ergothérapeutes et spécialistes de la mobilité. L'ophtalmologiste surveille la santé des yeux tandis qu'un généticien peut informer sur la transmission héréditaire de la maladie et les risques familiaux. De plus, des essais de nouveaux traitements, comme les implants de rétines artificielles et la thérapie génique, sont en développement.
Merci aux docteurs Chafik Keilani et Raphaël Atia, Chirurgiens ophtalmologistes au CHNO des Quinze-Vingts.